random
Simulate SimBiology model, adding variations by sampling error model
Syntax
Description
[
returns simulation results ynew,parameterEstimates]
= random(resultsObj)ynew with added noise using the
error model information specified by the
resultsObj.ErrorModelInfo property and estimated parameter
values parameterEstimates.
[
uses the specified ynew,parameterEstimates]
= random(resultsObj,data,dosing)data and dosing
information.
[
also applies the specified variants to each simulation.ynew,parameterEstimates]=
random(resultsObj,data,dosing,'Variants',v)
Note
The noise is only added to states that are responses which are the states
included in the responseMap input argument when you
called sbiofit or the ResponseMap
property of fitproblem. If there is a separate error model
for each response, the noise is added to each response separately using the
corresponding error model.
Examples
This example uses the yeast heterotrimeric G protein model and experimental data reported by [1]. For details about the model, see the Background section in Parameter Scanning, Parameter Estimation, and Sensitivity Analysis in the Yeast Heterotrimeric G Protein Cycle.
Load the G protein model.
sbioloadproject gproteinStore the experimental data containing the time course for the fraction of active G protein.
time = [10 30 60 110 210 300 450 600]'; GaFracExpt = [0.35 0.4 0.36 0.39 0.33 0.24 0.17 0.2]';
Create a groupedData object based on the experimental data.
tbl = table(time,GaFracExpt); grpData = groupedData(tbl);
Map the appropriate model component to the experimental data. In other words, indicate which species in the model corresponds to which response variable in the data. In this example, map the model parameter GaFrac to the experimental data variable GaFracExpt from grpData.
responseMap = 'GaFrac = GaFracExpt';Use an estimatedInfo object to define the model parameter kGd as a parameter to be estimated.
estimatedParam = estimatedInfo('kGd');Perform the parameter estimation. Use the name-value argument ErrorModel to specify the error model that adds error to simulation data.
fitResult = sbiofit(m1,grpData,responseMap,estimatedParam,ErrorModel="proportional");View the estimated parameter value of kGd.
fitResult.ParameterEstimates
ans=1×3 table
Name Estimate StandardError
_______ ________ _____________
{'kGd'} 0.10877 0.001397
Use the random method to retrieve the simulation data with added noise using the proportional error model which was specified by sbiofit. Note that the noise is added only to the response state, that is the GaFrac parameter.
[ynew,paramEstim] = random(fitResult);
Select the simulation data for the GaFrac parameter.
GaFracNew = select(ynew,{'Name','GaFrac'});Plot the simulation results.
plot(GaFracNew.Time,GaFracNew.Data)
hold on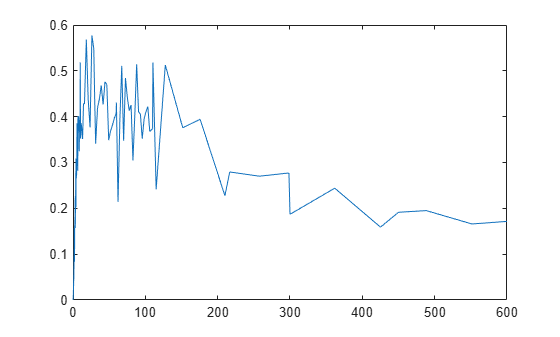
Plot the experimental data to compare it with the simulated data.
plot(time,GaFracExpt,'Color','k','Marker','o') legend('GaFracNew','GaFracExpt')
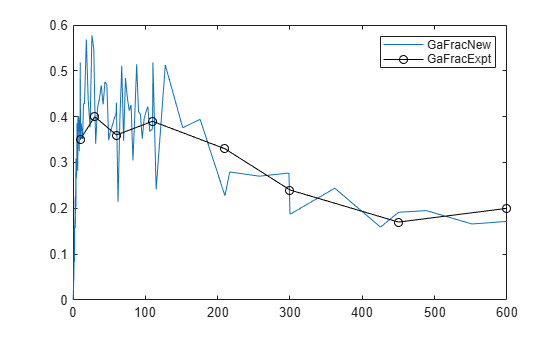
Input Arguments
Output Arguments
References
[1] Yi, T-M., Kitano, H., and Simon, M. (2003). A quantitative characterization of the yeast heterotrimeric G protein cycle. PNAS. 100, 10764–10769.
Version History
Introduced in R2014a